
从Nature Chemistry中解读理论计算方法:从头算分子动力学模拟(AIMD)
2024-10-30
1.论文简介

2.AIMD方法概述
从头算分子动力学模拟(Ab Initio Molecular Dynamics,AIMD)是一种结合量子化学和分子动力学的先进计算方法。它通过第一性原理(如密度泛函理论,DFT)直接描述电子和原子的相互作用,不依赖经验势函数,从而能够准确模拟材料中的化学反应和动态行为。
相比传统分子动力学(MD),AIMD在精度和适用范围上都有显著优势,特别适用于处理复杂体系和微观反应机制。
AIMD 的关键在于通过量子力学计算获取材料的电子结构,并结合分子动力学推演原子的运动。这使得AIMD能够在模拟过程中捕捉到体系的真实物理化学特性,无需假设额外的经验参数。
尽管AIMD计算精度高,但也面临计算成本较大的挑战。
为了确保AIMD模拟的高效运行,我们推荐使用以下服务器配置。这套配置能够提供强大的计算能力和稳定性,适合处理复杂的量子化学计算和大规模材料模拟:

这套配置能够提供高效的多核计算、快速数据读写以及稳定的散热效果,特别适合在大规模的AIMD计算中实现快速、稳定的性能表现。
然后本文将结合一项研究案例,通过AIMD模拟Li₃YCl₆中的Li⁺扩散行为,深入探讨其在温度变化下的扩散路径和相变机制,从而展示AIMD在揭示材料微观行为中的独特价值。
3.论文案例分析
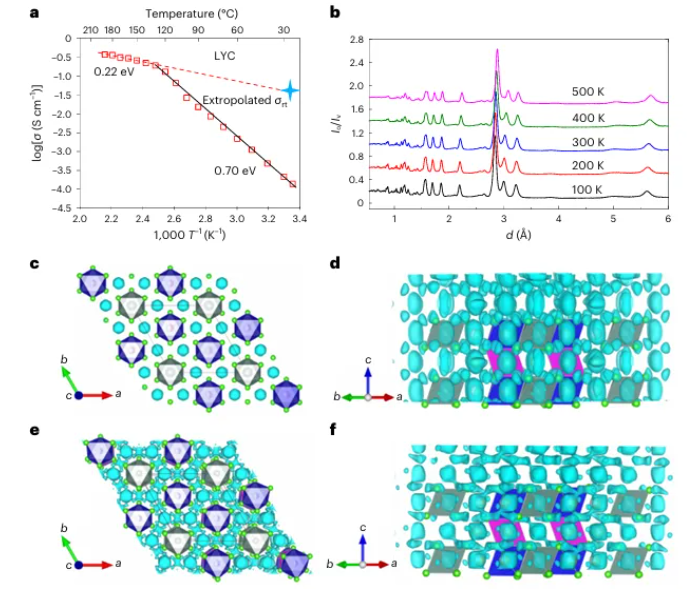
图2 | 使用不同温度下的ND模式生成的FDM可视化LYC的离子电导率和Li扩散路径。
a. LYC电导率的Arrhenius图。
b. 在不同温度下收集的LYC的ND模式。
c,d. 从Li₃YCl₆的ND模式在100K下生成的FDM,截断值分别为−0.035 fm Å⁻³(c)和−0.02 fm Å⁻³(d)。
e,f. 从Li₃YCl₆的ND模式在500K下生成的FDM,截断值分别为−0.03 fm Å⁻³(e)和−0.02 fm Å⁻³(f)。c轴方向的投影见补充图3和4。
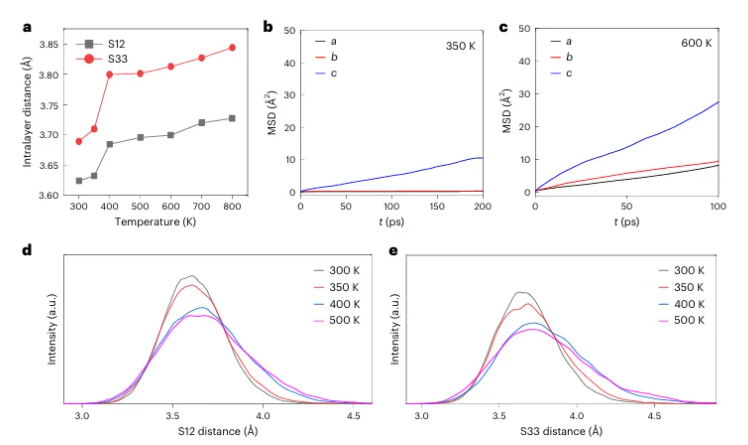
图4 | 不同温度下LYC的AIMD模拟。
a. S12和S13的平均层间Cl–Cl距离随温度的变化。
b,c. Li⁺在350K(b)和600K(c)下各方向的均方位移(MSD)。
d,e. 不同温度下的S12(d)和S33(e)层间Cl–Cl距离分布。
研究背景
本研究通过AIMD模拟,探究了Li₃YCl₆中的Li⁺扩散机制,特别是不同温度下的扩散行为和超离子相变现象。该材料是一种固态电解质,具有较好的化学和电化学稳定性,且在较高温度下表现出超离子导电性。理解其扩散机制对设计更高效的锂离子电池材料具有重要意义。
模拟方法与设置
AIMD的模拟设置包含温度梯度,以观察材料在不同温度下的Li⁺扩散路径变化。在低温条件下,Li⁺扩散主要集中在一维路径上;当温度升高时,材料内部的扩散路径逐渐拓展为二维。这一现象通过AIMD模拟中的路径分析得以清晰呈现。
关键发现
随着温度的升高,Li₃YCl₆中的Li⁺扩散路径从一维扩展为二维,这一扩散机制的转变被验证为一种超离子相变。AIMD的模拟结果显示了不同温度下的能量势垒变化,为实验中观测到的相变现象提供了理论支持。
实验结果对比
AIMD模拟的Li⁺扩散路径和能量势垒变化,与同步辐射X射线衍射等实验结果高度一致,进一步验证了AIMD在揭示材料微观行为中的有效性。在Li₃YCl₆材料的研究中,AIMD成功揭示了扩散路径的转变机制,量化了不同温度下的扩散系数,为材料设计提供了关键参数。
4.理论计算与材料设计
结合本研究中的Li₃YCl₆案例,AIMD可以用于材料性能优化、关键参数的定量分析,并扩展到其他材料设计。
1. 材料性能优化
AIMD模拟揭示了Li₃YCl₆中Li⁺扩散路径随温度从一维扩展至二维的变化机制,为实现更低温度下的超离子导电提供了思路。研究团队通过引入Br离子替代部分Cl离子,成功降低了Li₃YCl₄.₅Br₁.₅的超离子转变温度,从而在室温下提升了材料的导电性。
2. 定量分析
AIMD能够定量计算材料的扩散系数和能量势垒,帮助评估材料中的离子迁移阻力。通过长时间分子动力学模拟获取不同温度下的扩散路径和均方位移(MSD),研究者计算了激活能,揭示出高温下能量势垒显著降低,有助于Li⁺快速迁移。
3. 案例延伸
AIMD模拟还适用于其他材料的设计。通过进一步在Li₃GdCl₃Br₃中增加Br含量,AIMD预测其超离子转变温度可降至-10℃,并在室温下表现出更高的导电率。这一改性策略展示了AIMD在固态电解质设计中的广泛应用潜力。