
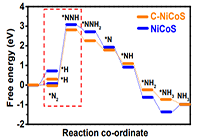
案例一:Predicted free energy evolution of N2 reduction on NiCoS/C and NiCoS
说明:During the process, it is found that free energy barrier to form the *N2 on NiCoS/C is lower than that on NiCoS, which means the absorption of N2 on the strong chemical coupled NiCoS/C is stronger than NiCoS, therefore, the NiCoS/C displays the better catalytic activity 85 for converting N2 to NH3.
来源文献:DOI: 10.1039/C9TA10142J
案例二:Predicted free energy evolution of NRR on 1T-MoS2
说明:The step for the reductive protonation of adsorbed N2 (*N2 → *NNH) shows the highest energy barrier of 0.59 eV without an external potential, indicating it is the potential-determining step (PDS) of the whole reaction.
来源文献:DOI: 10.1021/acsami.0c05204
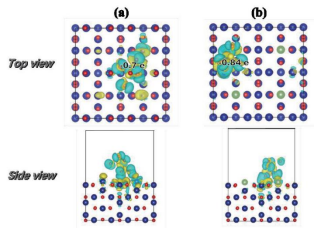
第一性原理计算的基本思想是将多个原子构成的体系看成是由多个电子和原子核组成的系统,并根据量子力学的基本原理对问题进行最大限度的“非经验性”处理。它只需要5个基本常数(m0,e,h,c,kB)就可以计算出体系的能量和电子结构等物理性质。它可以确定已知材料的结构和基础性质,并实现原子级别的精准控制,是现阶段解决实验理论问题和预测新材料结构性能的有力工具。并且,第一性原理计算不需要开展真实的实验,极大地节省了实验成本,现已被广泛应用于化学、物理、生命科学和材料学等领域。
适合的研究方向包括但不限于:催化、电池、半导体、金属材料、非金属材料、合金、纳米材料等
可以计算的体系包括但不限于:晶体、非晶、二维材料、表面、界面、固体等
常用软件:VASP,MS,CP2K,QE等
可以计算的内容包括但不限于:
材料的几何结构参数(如键长、键角、二面角、晶格常数、原子位置等)
材料的电子结构信息(如电荷密度、电荷差分密度、态密度、能带、费米能级、功函数、ELF等)
材料的光学性质(如介电常数等)
材料的力学性质(如弹性模量等)
材料的磁学性质(如磁导率等)
材料的晶格动力学性质(如声子谱等)
材料的表面性质(如吸附能,催化计算等)
复合材料的性质(异质结等内容)等等
