
(1)定义:在对接过程中受体与配体结构均不发生改变,仅改变分子的空间位置与姿态,是一种形状上的互补或相互作用。计算所用时间最短,适合于处理大分子之间的对接,如蛋白与蛋白的对接或蛋白与核酸的对接。
(2)作用:分析关键结合位点,主要考虑分子之间的契合程度。
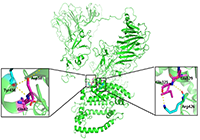
Fig. 6. Structural simulation of SARS-CoV-2 S-protein docking with human ACE2 molecule. Left panel: This area shows the hydrogen bond interaction between Tyr436 in S- protein and Asp38/Gln42 in ACE2. The relevant residues are presented in ball and stick representations. Right panel: This region shows the hydrogen bond interaction between Arg426 in S protein and Gln325/Glu329 in ACE2.
(3)说明:Analysis of the key binding sites of SARS-CoV-2 and ACE2 showed that SARS-CoV-2 S-protein Arg426 and Tyr436, respectively, combined with Gln325/Glu329 and Asp38/Gln42 of ACE2 through hydrogen bonds and formed an interaction interface with adjacent residues (Fig. 6), which is consistent with previous research results of Hao Pei team.
文章来源:DOI:10.1016/j.jff.2020.104016
分子动力学(Molecular Dynamics,MD)模拟是一套分子模拟方法,是研究凝聚态系统的有力工具。通过分子动力学模拟,研究者得到体系原子的运动轨迹,可观察到原子运动过程的各种微观细节。通过对研究体系的动态模拟,我们能够在分子水平上理解生物大分子的运动与生物功能、蛋白-小分子之间相互作用机理、纳米材料分子的自组装过程。分子动力学模拟是理论计算和实验方法的有力补充,广泛应用于物理、化学、材料科学和生物医药等领域。
适合的研究方向包括但不限于:生物、生化、医药、医学、药物、高分子、食品、材料、环境等
可以计算的体系包括但不限于:生物体系、蛋白质、核酸、多肽、药物分子、聚合物、小分子等
常用软件:gromacs,lammps,amber等
可以计算的内容包括但不限于:
蛋白三维模型搭建,如同源建模、从头建模等
分子对接,如蛋白质-小分子,核酸-小分子,小分子-小分子,蛋白-蛋白,蛋白-多肽,蛋白-核酸等
生物三维结构分析,如蛋白在不同pH、温度、电场下的三维结构变化等
动力学研究,如生物体系的弱相互作用分析、受体-配体组装过程、结合自由能分析,材料体系的高分子构象预测、材料与溶液界面性质,粗粒化模拟等
动力学后数据分析,如回旋半径(RMSF)、径向分布函数(RDF)、扩散系数、RMSD、各种能量分析、氢键数量分析、亲疏水性分析等
药物相关内容,如药物衍生物库设计、虚拟筛选、成药性预测、毒性分析、QSAR预测模型构建等
分子动力学(Molecular Dynamics,MD)模拟是一套分子模拟方法,是研究凝聚态系统的有力工具。通过分子动力学模拟,研究者得到体系原子的运动轨迹,可观察到原子运动过程的各种微观细节。通过对研究体系的动态模拟,我们能够在分子水平上理解生物大分子的运动与生物功能、蛋白-小分子之间相互作用机理、纳米材料分子的自组装过程。分子动力学模拟是理论计算和实验方法的有力补充,广泛应用于物理、化学、材料科学和生物医药等领域。
